Molecular modelling of crystal structures --
part 1
Calculations on structures from the CSD
In this excercise, you will retrieve structures from the CSD and make
working Cerius2 models out of those.
1. Estrone
How many structures of the steroid estrone are there in the CSD, and what
are the diferences between them?
To answer these questions, you will have to log in to the server at the
cmbi, and run the CSD interface.
Using the CSD interface
* log in at cmbi1.cmbi.kun.nl with the username/password you got earlier
from the CMBI system manager.
To log in, open a terminal window on your workstation (Desktop | Open Unix
Shell), and type
ssh -X cheminf.cmbi.ru.nl
in the window.
If you username here is different from the one you got
at the CMBI, you have to type
ssh -X username@cheminf.cmbi.ru.nl
where username is your username at the CMBI.
* Start the cheminformatics menu by typing xmenu & in the terminal window
* When the menu program has started, select ConQuest from the Cheminformatics menu.
After a few
seconds, the following window should appear:
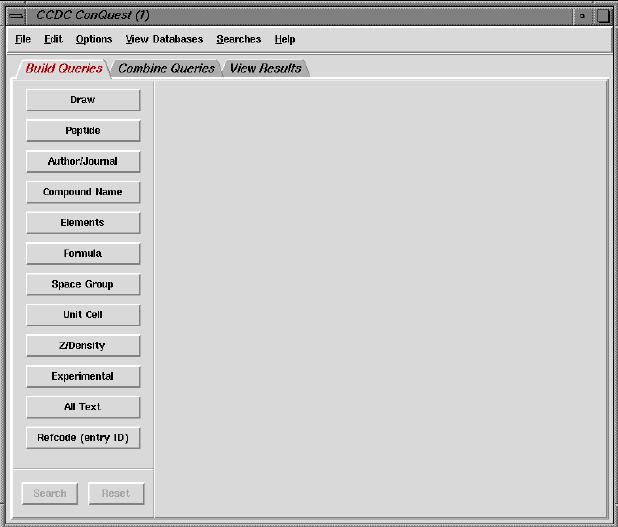
Find your own way through the interface, and get and save (in cif format)
all estrone structures. Remember, we want just estrone, not any of the
derivatives that are there as well in the database!
Transferring the structures
Now that you have the structures, get them to your workstation via the 'secure
copy' command, scp:
* in the cmbi1 terminal window, type
scp estron-data-file vstofchem17.sci.kun.nl:
(replacing estron-data-file by whatever you called your data file),
and provide your password as requested.
Note that there is a ":"-sign following vstofsgi.sci.kun.nl!
The file will now be copied to your local (VSC) home directory.
NB. If your local username is different from the one at the CMBI, type
scp estron-data-file local-username@vstofsgi.sci.kun.nl:
where local-username is your local user name.
Analysing the structures in Cerius2
You now have the data on the workstation, and can load them into
Cerius2, via File | load; set the file type to "CIF".
How many structures are there? Are they all different
polymorphs?
If a chemist would sketch an estrone molecule, the sketch would look
somewhat different from the molecules as they appear on the screen.
Name two important things which are missing in (some
of) the structures.
It may not always be easy to see whether two structure are the same, almost
the same, or different. The X-ray powder diffraction (XRPD) pattern may be
helpful then: if structures are (almost) the same, their XRPD patterns are
also. In Cerius2, you can calculate XRPD patterns, and compare
patterns from different models by subtracting one from the other.
In the card deck, go to the Analytical 1 card, as shown below.
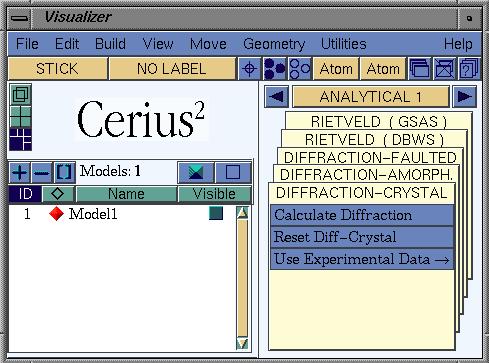
Calculate the XRPD pattern for the each structure by clicking
Calculate Diffraction, which opens a new window, and clicking
CALCULATE difraction pattern there.
Note: the calculation is done for the currently active structure.
To compare patterns, you can look at their difference pattern. To do so, use
the preferences for powder patterns (see the window below). Now, each time a
pattern is calculated, the difference with the
previous pattern is plotted as well.
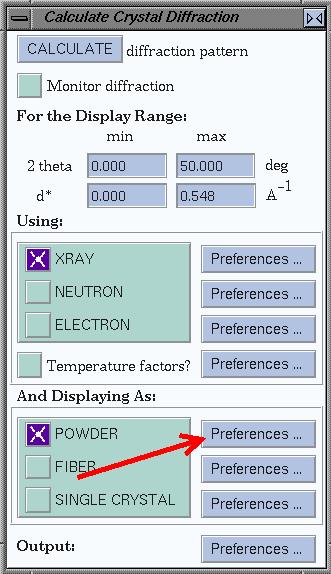
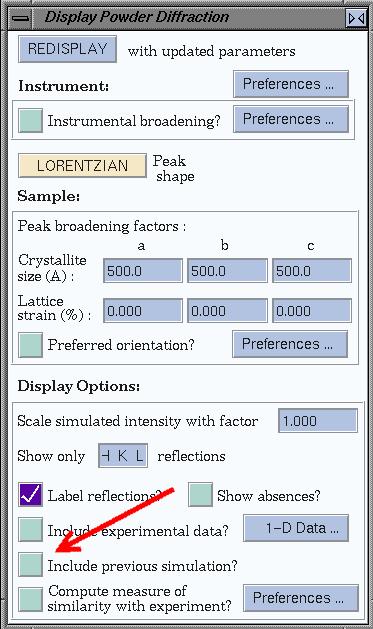
Compare all structures by their XRPD pattern. What are
your findings?
2. OD14
Get both structures of the steroid known as OD14 from the CSD. You
should find two polymorphs.
Give the crystallographic highlights of the polymorphs;
what is common, what not?
Clean up the structures by:
* 'unbuilding' the crystal (Card deck Builders1; Crystal
Building | Unbuild Crystal)
* using the 3D-Sketcher from the Build menu (not card deck)
to get the molecules right
* re-building the crystal
Save the structures to disk so you can use them later, using the .msi format.
Hydrogen bonds can be spotted automatically by Cerius2, via
Build (in the main Vidualizer window) | Edit H-Bonds....
Have a look at the hydrogen-bonding scheme of the polymorphs. Do they make
sense? How have they been influenced by your cleaning-up the structures?
The molecules have some interesting conformational differences. Let's look at
the energy difference between the conformations. To do so, make a new model
for each conformation. Load a force field (OFF SETUP | OPEN FORCE FIELD |
Load; choose the Dreiding2.21 force field), and
put atomic charges on the molecules. Save the structures for later use.
Wich are the main methods to assign atomic charges?
What are the most important differences? Why are Gasteiger charges
a practical choice in this case?
To get a realistic value for the energy, the energy should be calculated after
optimization of the geometry, and not on the structure as it is stored in
the data base. First make a copy of the structure by generating a new
model space (1), making the structure you want to copy also visible (2),
and set the arrangement of visible structures (3), as shown below. Then drag
the visible model into the empty model space (4).
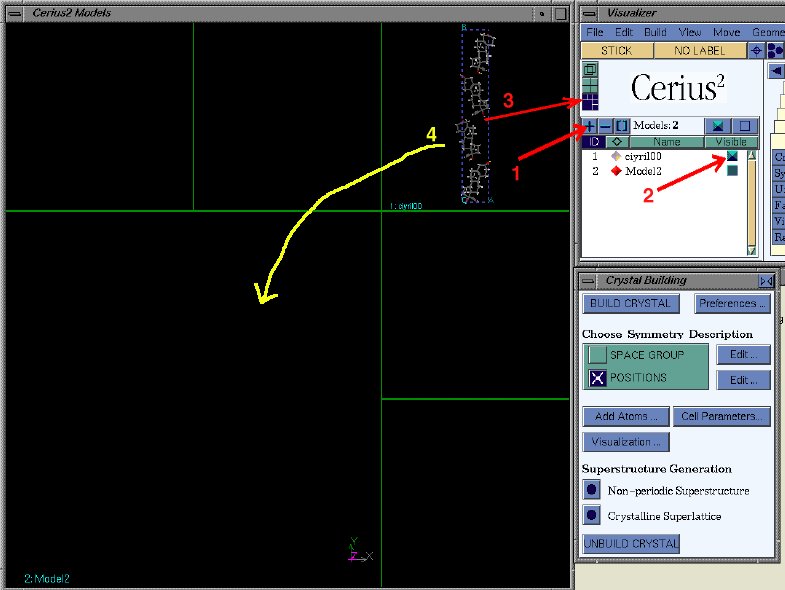
Make the original model not-visible again. Now unbuild the crystal structure,
so you get just the asymmetric unit. It contains two molecules (which are
not related by symmetry). Repeat the above procedure to copy a model, to
obtain another model of the asymmetric unit. Then delete one molecule from
each of the models, so you end up with a model of just one molecule, and
another model of only the other molecule.
To do the energy calculation, load the Dreiding force field: select the
OFF SETUP card, click Load, and pick DREIDING2.21. Then go to
the OFF METHODS card, and click Run. This will bring up a window
where you can calculate the structure's current energy, or do an energy
minimization.
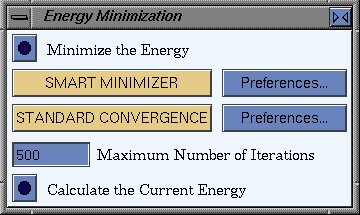
What are the absolute/relative energies of the two conformations? What happens
if you do not optimize the geometry?
Finally, we want to calculate the energy difference between the two
polymorphs. Be sure to handle the non-bonded interactions correctly, via the
OFF SETUP | OPEN FORCE FIELD | Energy Terms | van der Waals/Coulomb
windows in the card deck. Note that the
settings that you enter here, may be undone when you (re-) load the
force field, so always remember to do that first!
Also, note that the numbers you get from Cerius are in kcal per
mol unit cells!
So if there are N molecules per unit cell, you have to divide the
number by N in order to arrive at kcal/mol.
What is the problem with non-bonded interactions in large systems, and how
are these interactions handled best for 2/3D periodic systems?
What is the energy difference between the polymorphs?
You can compare the optimized structures with the original ones from the CSD
by overlaying both in the models window. Is the optimized structure
still similar to the original? You can also compare their XRPD patterns,
like you did for estrone. How similar are the patterns?
Once you have done all this, get back to the
main course page.